
胶束电解质实现对水系锌钒电池的局部水限制
研究简介
高反应性水引起的级联故障,包括钒溶解、质子嵌入、析氢反应和界面副反应,限制了钒基水系锌离子电池的可回收性。这些故障在低电流密度(<0.5Ag−1)下更为严重。目前对电解质优化的研究可以稳定锌负极,但忽略了钒基正极。本文从钒基正极的角度出发,使用表面活性剂十六烷基三甲基溴化铵(CTAB)开发了一种胶束电解质,其中水被局部限制,Br−重组Zn2+溶剂化,共同抑制了水引起的级联故障。同时,静电相互作用使CTA⁺能够插入V─O层(形成扩展间距的正极(CTA,Ca)VO)并产生正极表面双电层,从而增强了伪电容以抵消水限制引起的动力学损失。此外,循环诱导的CTA+降解参与了固态电解质界面相(CEI/SEI)的形成,从而提供进一步有效的正极/负极界面保护。胶束电解质平衡了水限制和电荷转移,实现了突破性的全电池性能:在0.1/0.2/4.0Ag−1(25°C)下经过150/300/17700次循环后,电荷保留率分别为93.57%/98.78%/82.17%;在0.1Ag−1(−20°C)下经过420次循环后,电荷保留率分别为99.77%。该胶束电解质策略可扩展到其他钒基正极(例如NaVO、BaVO)、准固态电池和无负极电池,为电解质设计提供了可行的范例。
图文导读
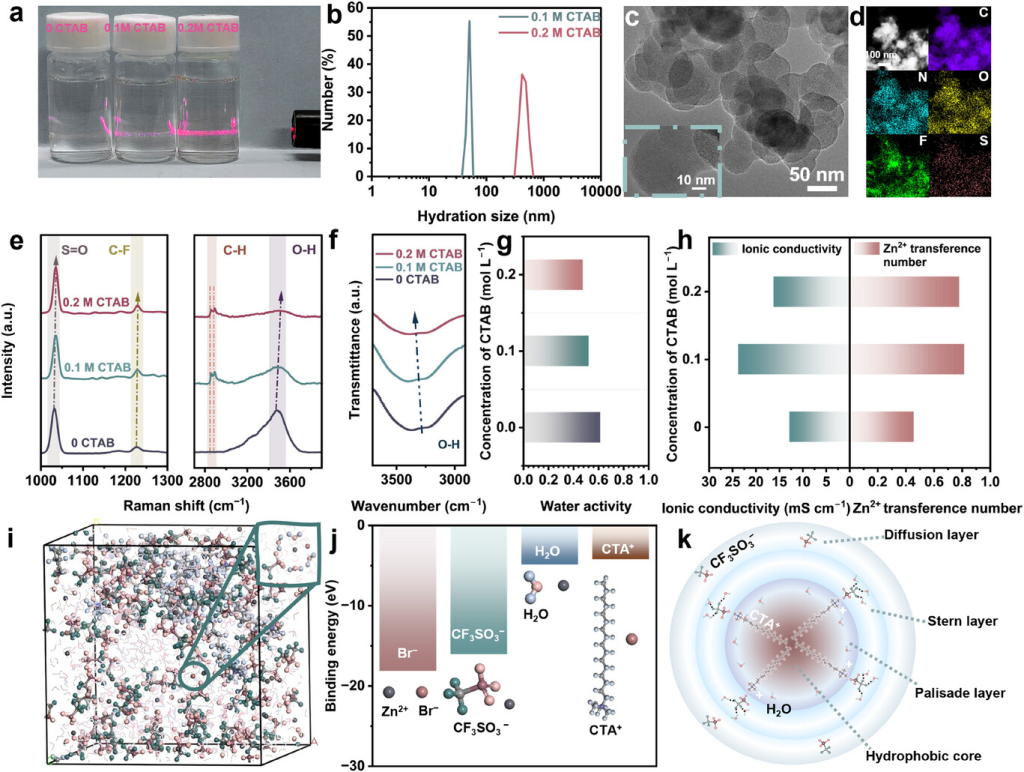
图1. 胶束电解质的设计概念和特性。a)电解质的廷德尔效应(0CTAB、0.1mCTAB和0.2mCTAB)。b)电解质中胶束颗粒尺寸分布的DLS分析(0.1mCTAB和0.2MCTAB)。c)TEM图像和 d)0.1mCTAB的EDS映射。e)拉曼光谱、f)FTIR光谱、g)25°C下的水活度和h)电解质(0CTAB、0.1mCTAB和0.2MCTAB)的离子电导率和Zn2+迁移数。 i)0.1mCTAB的MD模拟快照和水合Zn2+的代表性溶剂化结构。j)Zn2+与不同离子的结合能。k)0.1mCTAB中CTA+胶束结构示意图。
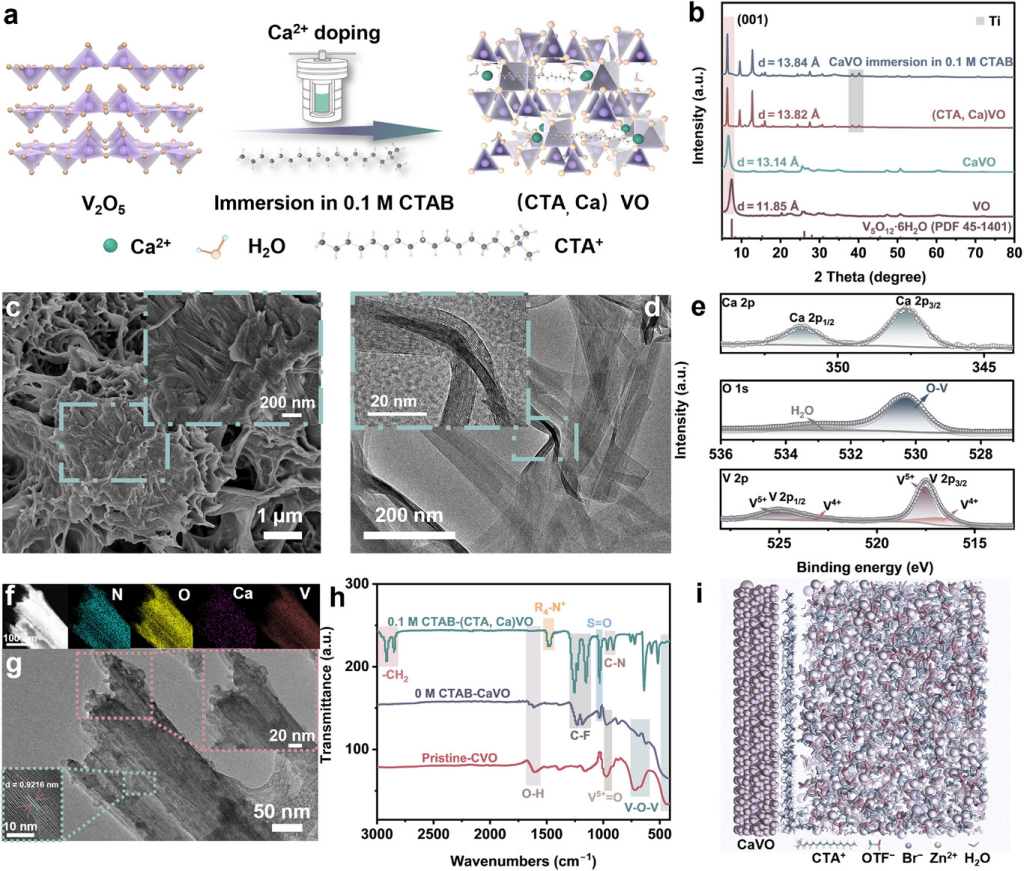
图2.正极特性和胶束电解质对阴极的影响。a)(CTA,Ca)VO合成示意图。b)浸入0.1MCTAB中的VO、CaVO、(CTA,Ca)VO和CaVO的XRD图案。c)CaVO的SEM和d)HRTEM图。e)CaVO的Ca2p、O1s和V2p的XPS光谱。f)浸入0.1MCTAB中的CaVO样品的EDS映射和g)TEM图。h)在0M/0.1MCTAB中循环一次后CaVO和CaVO的FTIR光谱。i)CaVO-电解质(0.1MCTAB)界面处的MD模拟快照。
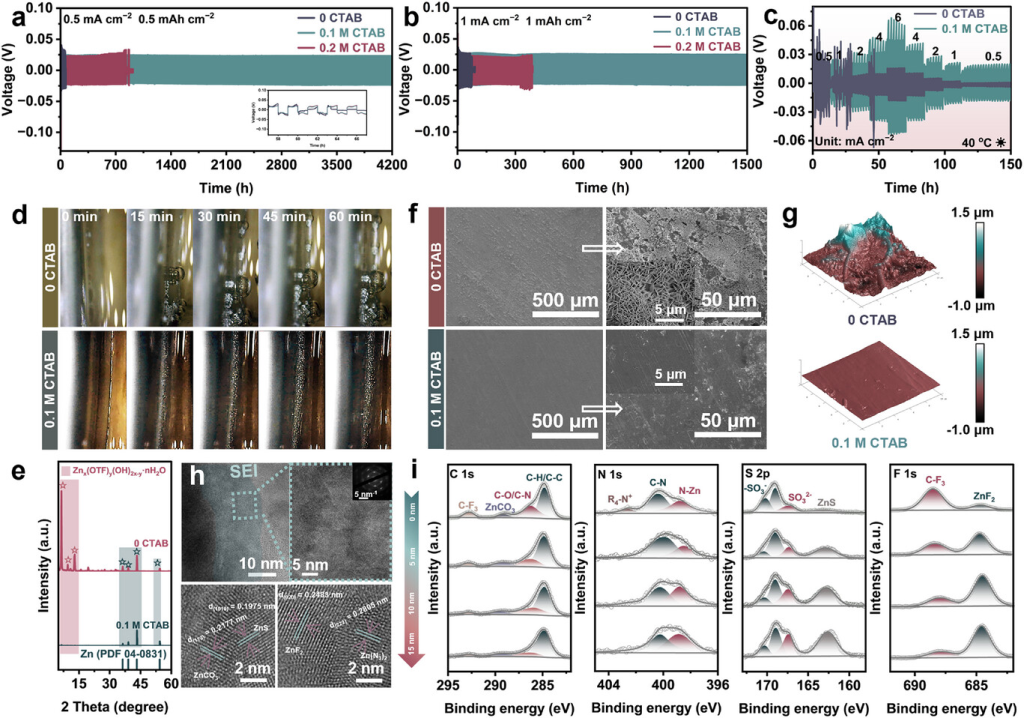
图3. CTAB添加剂对锌负极电化学性能和界面的影响。a)在0.5mAcm−2和0.5mAhcm−2下使用0CTAB和0.1MCTAB的Zn||Zn电池的长期剥离/电镀稳定性,以及b)1mAcm−2和1mAhcm−2。c)在40°C和不同电流密度下使用0CTAB和0.1MCTAB的Zn||Zn电池的倍率性能。d)在6mAcm−2下0CTAB和0.1MCTAB中Zn沉积的光学显微照片。e)在0CTAB和0.1MCTAB电解质中循环后锌电极的XRD。f)SEM和g)在0CTAB和0.1MCTAB中循环后锌电极表面的AFM图。g)在0CTAB和0.1MCTAB中循环后锌负极的粗糙度和沿箭头所示方向的高度分布。h)HRTEM图和i)在0.1MCTAB中循环后锌电极表面的C1s、N1s、S2p和F1s的XPS深度剖面。
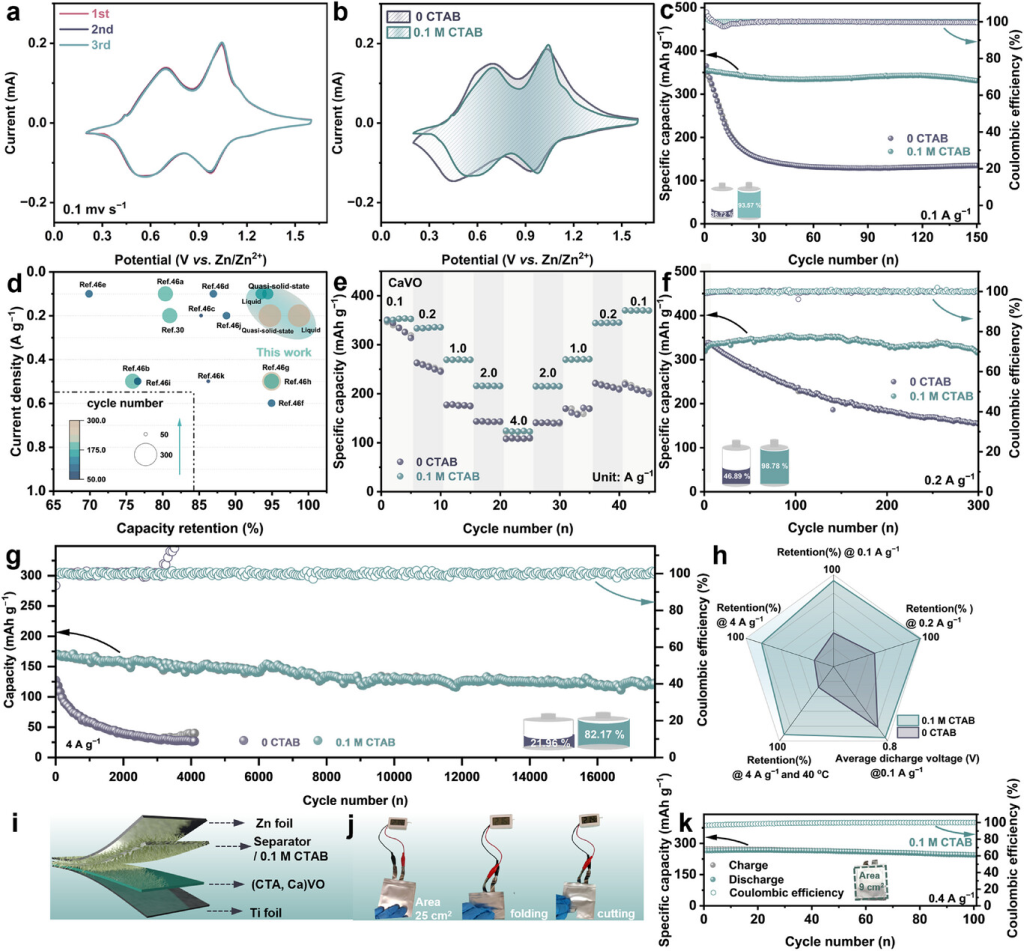
图4. Zn||(CTA,Ca)VO全电池的电化学性能。a)0.1mCTAB的Zn||(CTA,Ca)VO电池和b)0CTAB的Zn||CaVO电池在0.1mVs−1下的CV曲线。c)0.1mCTAB的Zn||(CTA,Ca)VO电池与0CTAB的Zn||CaVO电池在0.1Ag−1下的循环性能。d)低电流密度条件下循环稳定性的比较分析。e)0.1M CTAB的Zn||(CTA,Ca)VO电池和0CTAB Zn||CaVO电池在不同电流密度下的倍率性能。f)0.2Ag−1、g)4Ag−1下的长期循环稳定性,以及 h)Zn||(CTA,Ca)VO和Zn||CaVO电池电化学性能的总体比较。i)软包液体电池的示意图,j)在不同状态下为温度计供电的液体Zn||(CTA,Ca)VO软包电池的光学图。k)Zn||(CTA,Ca)VO软包电池在0.4Ag−1下的循环性能。
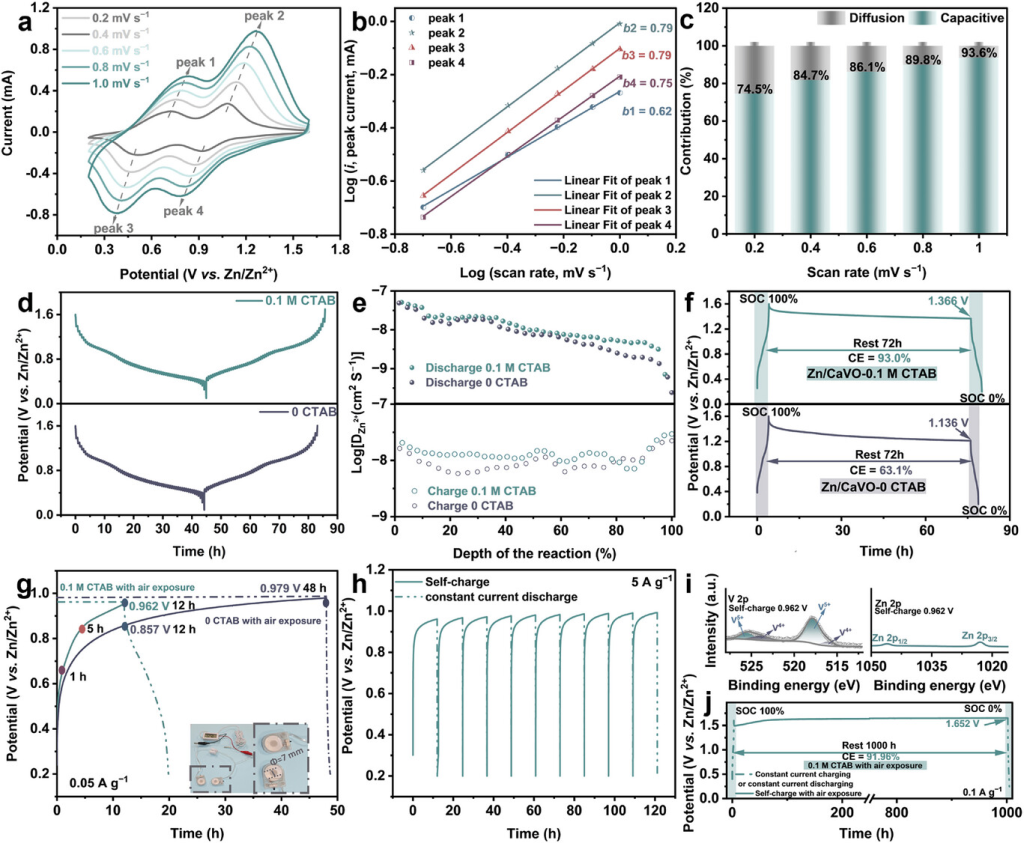
图5.Zn||(CTA,Ca)VO全电池的动力学和自充电性能。a)在不同扫描速率下使用0.1mCTAB的Zn||(CTA,Ca)VO电池的CV曲线。b)峰值电流与扫描速率的对数相关性。c)电容/扩散电荷存储贡献的量化。d)Zn||(CTA,Ca)VO(0.1mCTAB)和Zn||CaVO电池(0mCTAB)的GITT曲线。e)比较Zn2+扩散动力学。f)自放电测试。g)在0.05Ag−1下进行自充电测试,其中纽扣型Zn||(CTA,Ca)VO自充电电池为温度计供电的光学图。h)在5Ag−1下进行自充电测试。i)自充电(CTA,Ca)VO阴极中Zn2p和V2p的XPS光谱。j)Zn||(CTA,Ca)VO自充电电池的自放电测试。
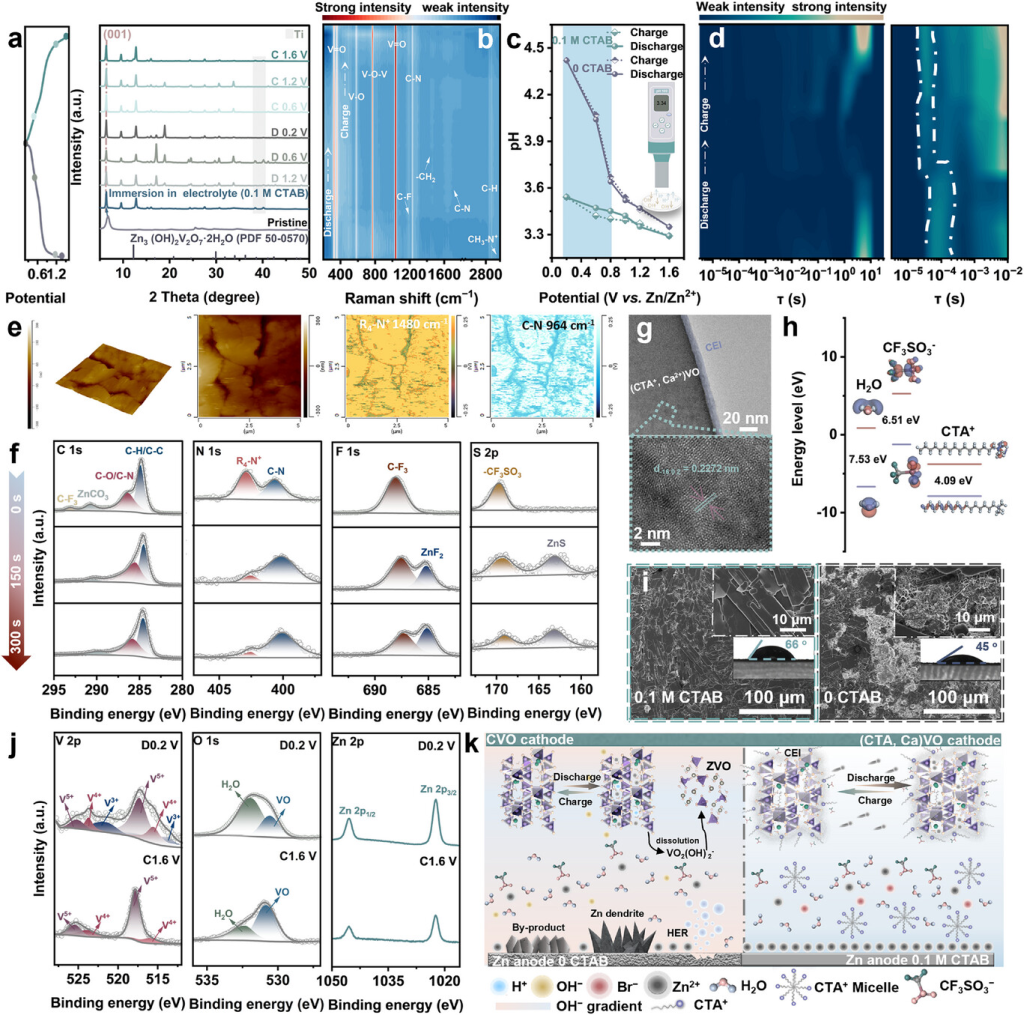
图6.Zn||(CTA,Ca)VO电池中电化学演变的多模态表征。a)不同充电/放电状态下(CTA,Ca)VO的原位XRD。b)原位拉曼光谱监测电化学循环过程中的动态结构演变;c)正极/电解质界面处的原位pH测量。d)(CTA,Ca)VO正极在0.1mCTAB中的放电/充电过程的DRT曲线。e)循环(CTA,Ca)VO正极的R4-N+和C─N振动的AFM形貌图与纳米红外重叠。f)循环(CTA,Ca)VO正极的C1s、N1s、S2p和F1s核心能级的XPS深度曲线。g)循环后CEI层和正极的HR-TEM图。h)计算的前沿分子轨道能级和相应的带隙。i)(CTA,Ca)VO与CaVO正极后循环的SEM图和接触角测量。j)循环过程中V2p、O1s和Zn2p的XPS光谱演变。k)电化学循环过程中结构演变和离子传输机制示意图。
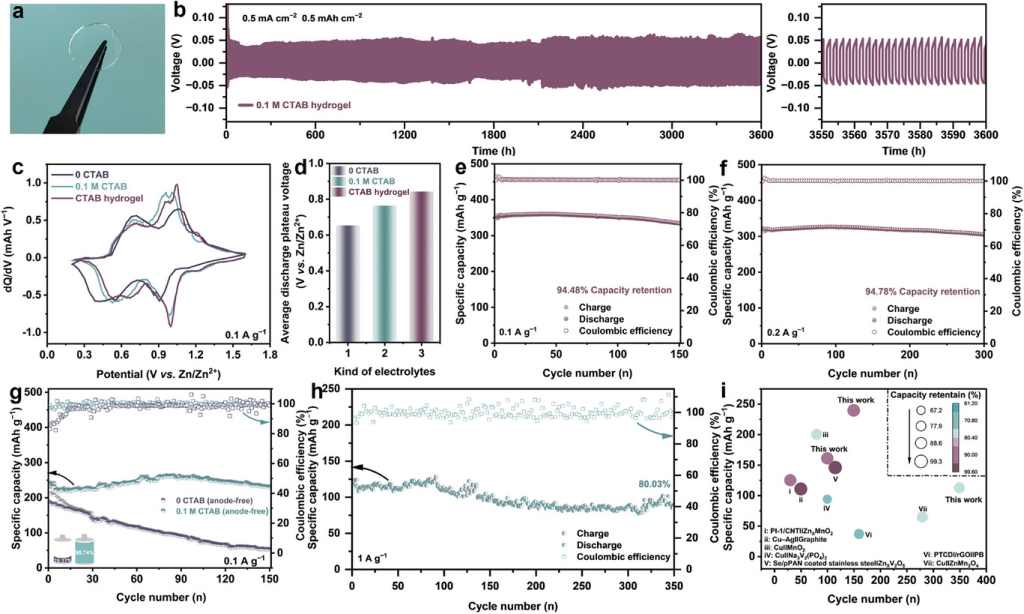
图7. 胶束电解质的扩展应用评估测试。a)CTAB水凝胶的光学图。b)使用CTAB水凝胶在0.5mAcm−2和0.5mAhcm−2下Zn||Zn对称电池的长期剥离/电镀稳定性。c)DQ/dV曲线和d)电解质中全电池在0.1Ag−1下的平均放电电压。CTAB水凝胶中Zn||(CTA,Ca)VO在e)0.1Ag−1和f)0.2Ag−1下的循环性能。0.1MCTAB电解质中Cu||(CTA,Ca)VO无负极全电池在g)0.1Ag−1和h)1.0Ag−1下的循环性能。i)我们的工作与其他报道的无阳极电池的比较。
研究结论
本研究开发了一种三氟乙酸(TFA)驱动的选择性蚀刻策略,以设计具有优先暴露的(002)晶面的ZnMP阳极,解决了限制AZIB实际部署的基本挑战。TFA的双功能机制,其中H+离子优先蚀刻高能面,而CF3COO−阴离子选择性保护(002)平面,产生了独特的阶梯式六边形形态,从根本上改变了ZnMP电极的电化学行为。这些改进转化为卓越的电化学稳定性,对称电池在电流密度为1mAcm−2和面积容量为0.5mAhcm−2下循环超过1000小时。此外,在大型4×3cm²软包电池中的成功演示,在10Ag−1下经过1000次循环后实现79.8%的容量保持率,验证了该方法的实际可行性。通过密度泛函理论(DFT)计算和相场模型进行的理论验证,为(002)富集表面带来的优先离子吸附机制和增强的电场均匀性提供了基础性的见解。这一机制理解为颗粒电极的晶体学控制建立了一个可推广的框架,其应用范围可拓展至锌体系之外。该表面工程策略能够同时解决互连表面失效、析氢、枝晶生长和副产物积累等问题,同时保留粉末电极的高表面积优势,为高性能AZIB用于固定式储能提供了一条切实可行的途径,有助于更广泛地向可持续、安全的储能技术过渡。