
阳离子-阴离子协同化学实现局部贫水和高电位差界面工程实现稳定的无析氢锌金属负极
研究简介
镀锌过程中的析氢反应(HER)和不受控制的沉积动力学严重破坏了锌金属负极的可逆性。内部亥姆霍兹平面及其形成的固体电解质界面(SEI)对深度调控析氢反应(HER)至关重要。我们报道了一种局部贫水、高电位梯度界面设计,该界面设计使用三氟乙酸铥作为电解质添加剂。三氟乙酸根阴离子在锌表面形成一层富含阴离子的层,作为质子阻挡屏障,并诱导局部缺水界面。该层与原位富含ZnF2的SEI相共同抑制了水分解引起的析氢反应(HER)。同时,Tm3+介导的双电层增强了Zn2+的传输,并引导以(002)取向为主的六方排列的锌生长。结果表明,Zn∥Zn对称电池在20mAcm-2/5mAhcm-2的电流密度下几乎不产氢,且稳定性能超过1000小时;而Zn∥聚苯胺软包电池在1000次循环后仍能保持82.3%的容量且不发生膨胀。这种阳离子-阴离子协同作用策略为实现无产氢的锌金属负极提供了一种途径。
图文导读
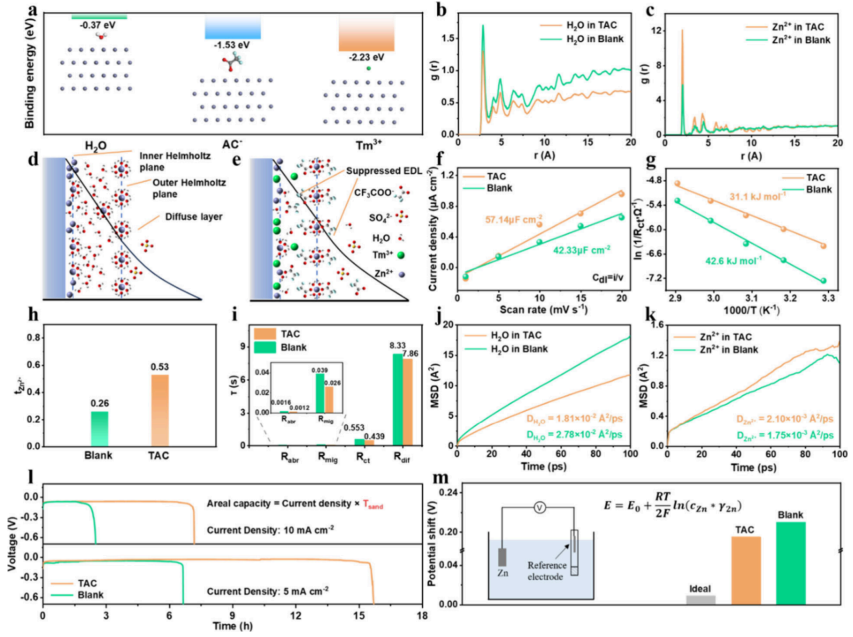
图1. EDL对Zn2+动力学的影响。(a)H2O和AC–在Zn(002)表面的吸附模式和相应的能量。(b)Zn2+和(c)H2O在Zn电极表面的密度分布图。(d)空白和(e)TAC电解液扩散层的EDL模型示意图。(f)空白和TAC电解液中对称电池的EDL电容。(g)空白和TAC电解液中的活化能。(h)Zn2+的迁移率。(i)在5mAcm–2/5mAhcm–2下循环20次后对Zn对称电池进行的DRT分析。(j)空白和(k)TAC电解液中Zn2+和H2O的MSD。(l)电流密度为5mAcm–2和10mAcm–2时Zn对称电池的恒电流电压曲线。(m)Zn2+/Zn的平衡电位。
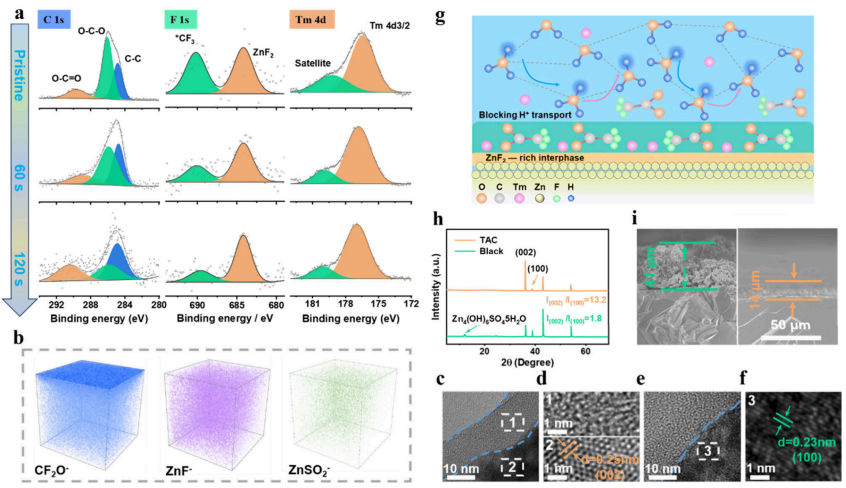
图2.含或不含TAC的TAC电解液中SEI的结构表征。(a)在Ar+溅射0、60和120秒后,TAC电解液中Zn负极的C1s、F1s和Tm4dXPS光谱。(b)TAC电解液中循环Zn金属表面各种物质的3DTOF-SIMS映射。(e,f)空白和(c,d)TAC电解液体系中Zn表面层状结构SEI的HRTEM图。(g)TAC添加剂在Zn负极界面的作用机理示意图。(h)空白和TAC电解液中Zn负极的XRD。(i)20次循环后的横截面SEM图。
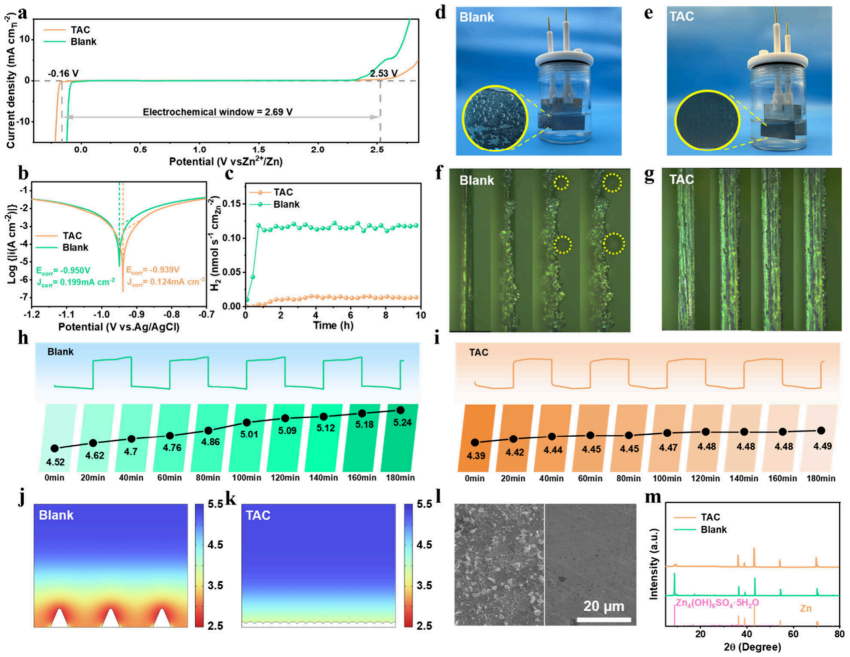
图3.抑制HER并稳定电解质pH值。(a)扫描速率为1mV/s时惰性(Ti)电极上的电化学稳定窗口。(b)Zn负极的线性极化曲线,以Ag/AgCl电极为参比电极。(c)通过OEMS测量的镀锌过程中的HER通量。在(d)空白和(e)TAC电解质中以7mAcm–2/7mAhcm–2循环20次后Zn∥Zn对称电极的形态表征。在(f)空白和(g)TAC电解质中以1mAcm–2在不同时间的Zn∥Cu的原位光学显微镜可视化。在(h)空白和(i)TAC电解质中以9mAcm–2循环的Zn∥Zn对称电池的原位pH演变。在(j)空白和(k)TAC体系中模拟Zn电极界面处的pH分布。在(l)空白和(m)TAC电解质中自腐蚀7天后的Zn表面的SEM图和XRD。
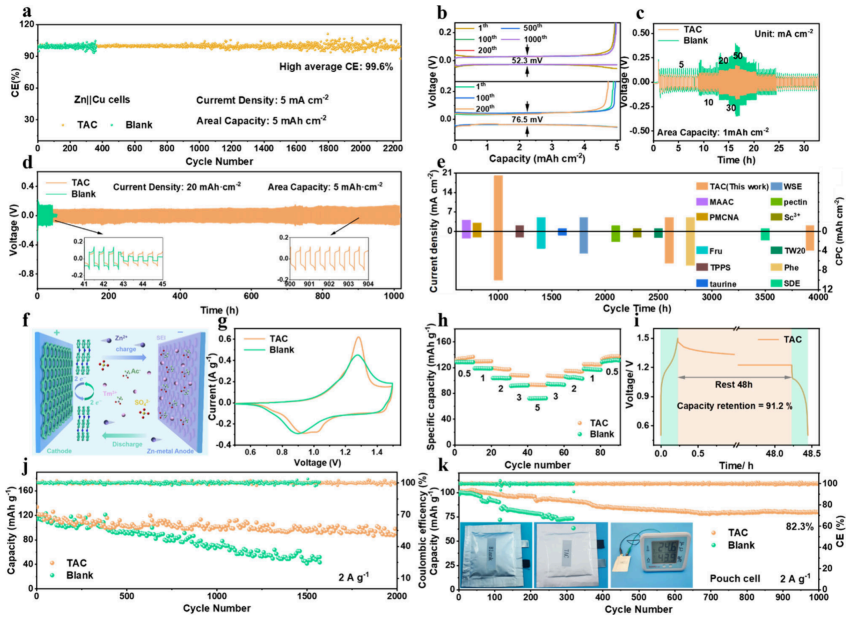
图4.电化学性能评估。(a,b)不同电解液中Zn∥Cu非对称半电池的CE和代表性电压曲线。(c)不同电流密度下Zn∥Zn对称电池的倍率性能。(d)20mAcm–2、5mAhcm–2下Zn∥Zn对称电池的电压曲线。(e)本工作与其他报道中Zn∥Zn电池的CPC比较。(f)Zn∥PANI全电池示意图。(g)扫描速率为1mVs–1时,Zn∥PANI纽扣电池在不同电解液中的CV曲线。(h)倍率性能。(i)Zn∥PANI纽扣电池静置48小时后的自放电测试。(j)不同电解液中2Ag–1电流密度下Zn∥PANI纽扣电池的长期循环稳定性。(k)Zn∥PANI软包电池在2Ag–1下的循环性能。插图:Zn∥PANI软包电池循环后的照片。
研究结论
本研究开发了一种新颖的界面工程策略,通过加入三氟乙酸根(TAC)作为电解液添加剂,在锌金属负极上营造局部贫水、高电势差环境。该方法通过阻断质子经富含阴离子层的传输,并结合局部贫水环境和原位SEI层来缓解水分解,从而有效抑制了电解液析出(HER)。此外,Tm3+离子的持续吸附压缩了扩散层的厚度,显著增大了OHP和本体电解液之间的电势梯度,进而加速了Zn2+离子的传输动力学。由此产生的Zn∥Zn对称电池利用Tm3+和三氟乙酸根离子之间的协同作用,在20mAcm-2/5mAhcm-2下实现了超过1000小时的长期稳定性。为了证明其实际应用价值,我们组装的Zn∥PANI软包电池在1000次循环后仍保留了82.3%的容量,且未出现任何明显的鼓包现象。这项研究凸显了阳离子-阴离子协同化学在设计新型电解质添加剂以抑制电解液析出(HER)和促进锌均匀沉积方面的潜力,为高性能AZIB的开发开辟了新的途径。