
通过空间和竞争性溶剂配位调控松散Mg2+溶剂化结构用于快速充电镁电池
研究简介
镁电池因其高安全性、高成本效益和丰富的资源,作为下一代储能技术正受到越来越多的关注。然而,由于电极/电解液界面处Mg2+传输动力学迟缓,镁电池的发展仍然受到限制。在此,我们提出了一种电解液设计策略,通过将四氢呋喃(THF)作为共溶剂引入硼酸盐基电解液MgB(hfip)4中的二甲氧基乙烷(DME)溶液中来调节Mg2+的溶剂化结构。THF选自一系列线性和环状醚,其介电常数和供体数与DME相当,但其环状结构引入了空间位阻,从而诱导与Mg2+的竞争配位。这种竞争削弱了Mg2+与溶剂的相互作用,导致溶剂化结构更不稳定,脱溶剂动力学增强。结果表明,采用优化的MBF/1D1T电解液(DME:THF=1:1,v:v)的Mg||Mg电池在10mAcm−2时Mg沉积/剥离过电位显著降低至120mV(使用MBF/DME时在8mAcm−2时为316mV),同时具有超过1200小时的出色循环稳定性。此外,在MBF/1D1T存在下,代表性硫化物正极如CuS和VS4表现出更快的活化速度和更好的高倍率性能。
图文导读
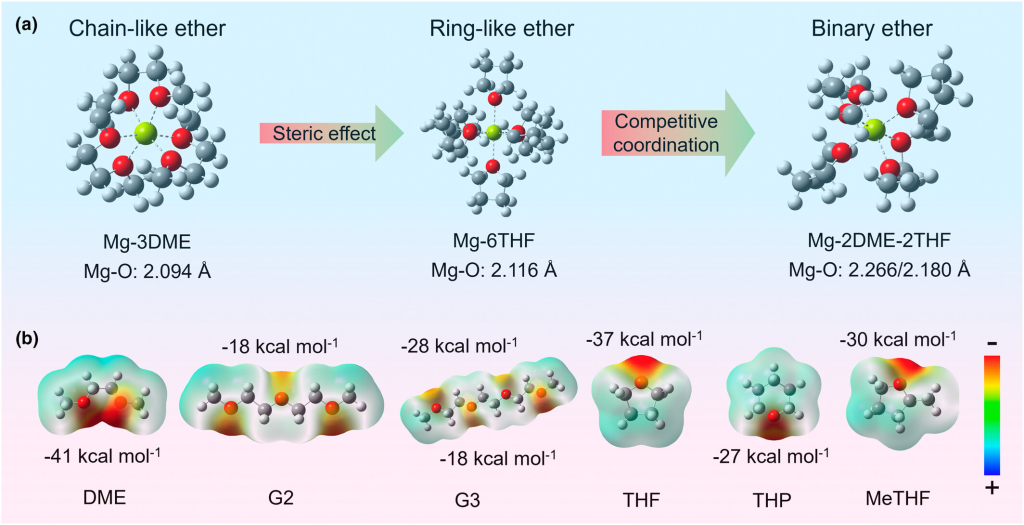
图1.a)通过DFT优化的Mg2+溶剂化结构和b)DME、G2、G3、THF、THP和MeTHF分子的表面静电势。
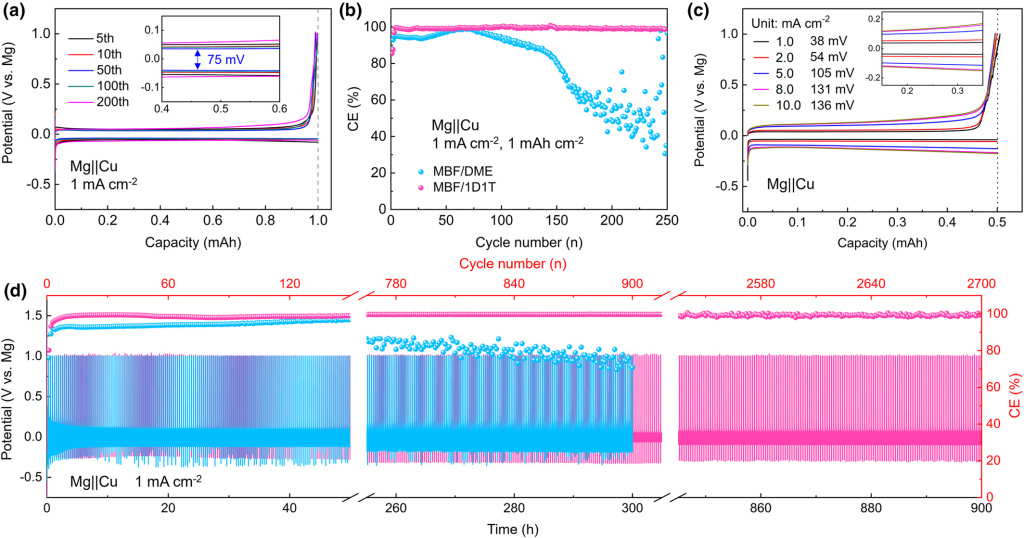
图2.a)使用MBF/DME或MBF/1D1T 电解液在1mAcm−2和1mAhcm−2下的Mg||Cu非对称电池的电压曲线和b)CE。c)使用MBF/1D1T电解液在1至10mAcm−2电流密度范围内的Mg||Cu非对称电池的电压曲线。d)使用MBF/DME或MBF/1D1T 电解液在1mAcm−2下的Mg||Cu非对称电池的循环性能。
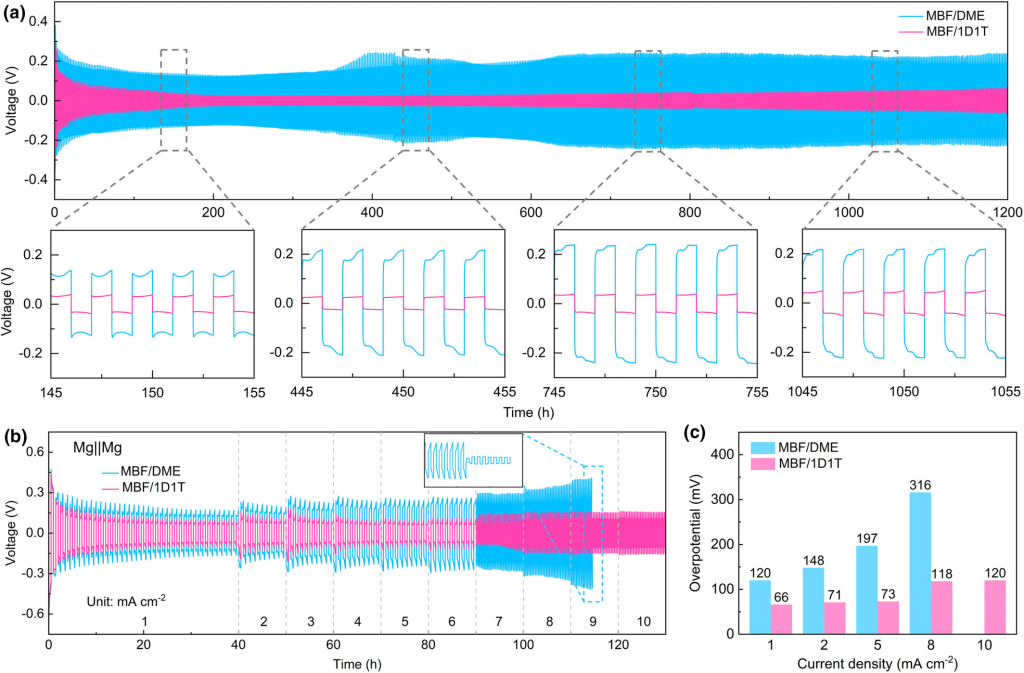
图3.a)使用MBF/DME和MBF/1D1T 电解液时0.5mAcm−2和0.5mAhcm−2下Mg||Mg对称电池的电压曲线。b)使用MBF/DME和MBF/1D1T 电解液时Mg||Mg对称电池的倍率性能和c)相应的过电位。
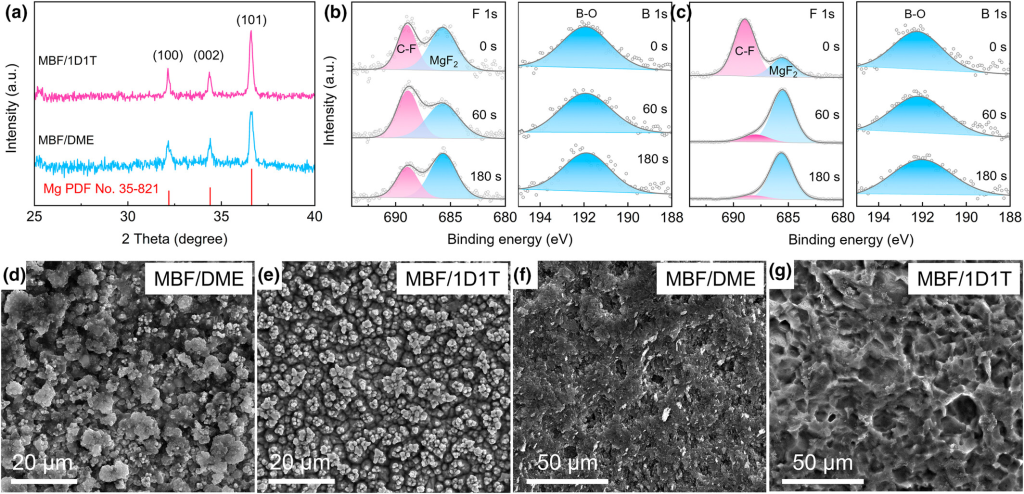
图4.a)MBF/DME和MBF/1D1T中Cu箔上Mg沉积物的XRD。b、d)MBF/DME和c、e)MBF/1D1T中Cu箔上Mg沉积物的XPS(F1s和B1s)和SEM图。f)MBF/DME和g)MBF/1D1T中50次循环后Mg金属表面的SEM图。
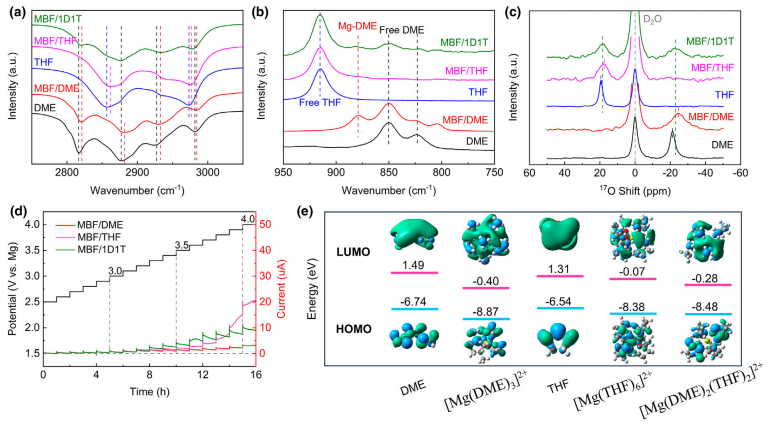
图5.a)FT-IR、b)拉曼和c)不同溶剂和电解液的17ONMR数据。d)使用不同电解液的Mg||Al电池的计时电流曲线,相对于Mg从2.5到4.0V测量。e)计算DME、THF和溶剂化Mg离子([Mg(DME)3]2+、[Mg(THF)6]2+和[Mg(DME)2(THF)2]2+)的HOMO和LUMO能级。
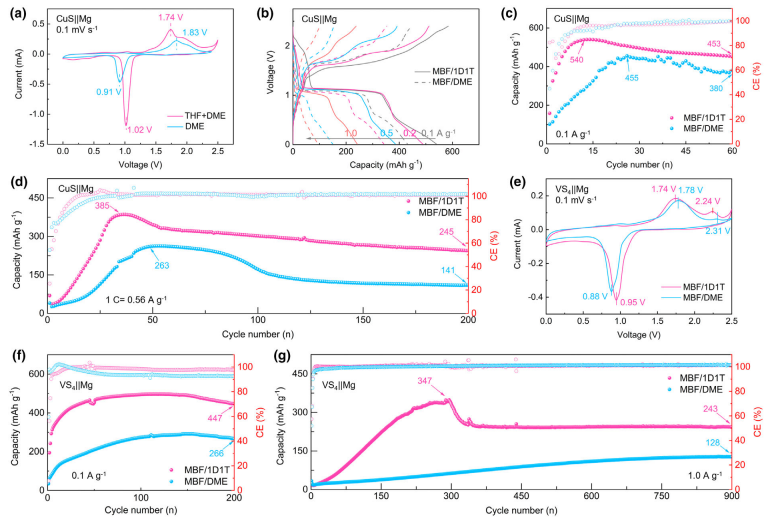
图6. a)0.1mVs-1时的CV曲线;b)不同电流密度下的充放电曲线;以及MBF/DME和MBF/1D1T电解液中Mg||CuS电池在c)0.1Ag-1和d)0.56Ag-1时的循环性能。e)使用MBF/DME和MBF/1D1T电解液的Mg||VS4电池在0.1mVs-1时的CV曲线。使用MBF/DME和MBF/1D1T电解液时Mg||VS4电池在f)0.1Ag-1和g)0.56Ag-1时的循环性能。
研究结论
通过将THF(一种介电常数和供体数与DME相当的醚溶剂)加入MBF/DME电解液中,开发了一种混合溶剂电解液策略。空间位阻的THF分子与Mg2+竞争性配位,从而削弱了整体的Mg-溶剂相互作用。全面的光谱表征,包括拉曼、FT-IR和17ONMR,证实了在定制的MBF/1D1T电解质中形成了弱配位溶剂化结构。这种优化的溶剂化环境显着改善了与镁金属的界面性能,证据是1mAcm−2时沉积过电位超低至38mV,明显低于MBF/DME 电解液中观察到的179mV,10mAcm−2时仅为136mV。此外,采用MBF/1D1T的Mg||Cu和Mg||Mg电池表现出了出色的循环稳定性,分别稳定运行了900和1200小时,超过了MBF/DME对应电池的200和300小时。此外,与MBF/DME相比,MBF/1D1F电解液可实现更均匀的Mg沉积,表明界面稳定性得到改善。当应用于硫化物基正极材料时,MBF/1D1T能够缩短活化过程并提高倍率性能。值得注意的是,与搭配MBF/DME的正极相比,CuS和VS4正极在1.0Ag−1时的容量分别提高了约2.3倍和2.7倍。这些发现凸显了有效的竞争协调策略在调节多价Mg2+溶剂化结构方面的应用。