
建立场流竞争模型揭示非单调界面Li+动态过程稳定高压正极-电解质界面
建立场流竞争模型揭示非单调界面Li+动态过程稳定高压正极-电解质界面
研究简介
揭示界面溶剂化结构的演变是解锁高性能锂离子电池的关键前沿。虽然本体电解质的溶剂化化学已被广泛表征,但界面物质在电场和浓度场下的动态行为及其在正极-电解质界面相(CEI)形成中的控制作用仍不清楚。本文,我们利用原位电化学光谱揭示了界面Li+粒子数的多阶段非单调演变,其呈现“富集-耗尽-再富集”的趋势。我们引入了一个场流竞争调控模型,其中界面电场驱动Li+迁移远离界面,而脱锂诱导的Li+流入则补充了界面浓度。该机制框架将界面溶剂化构型与操作参数(场强、电流密度、溶剂极性)关联起来,并最终通过相控溶剂化阶段决定CEI的组成。利用这些见解,我们开发了一种电位调制活化方案,以绕过有害的Li+/阴离子耗尽相,从而构建出机械性能稳定的CEI。该方法显著提高了高压正极的循环稳定性。通过揭示动态界面溶剂化工程的原理,这项工作为先进储能系统中电极/电解质界面的合理设计奠定了基础。
图文导读
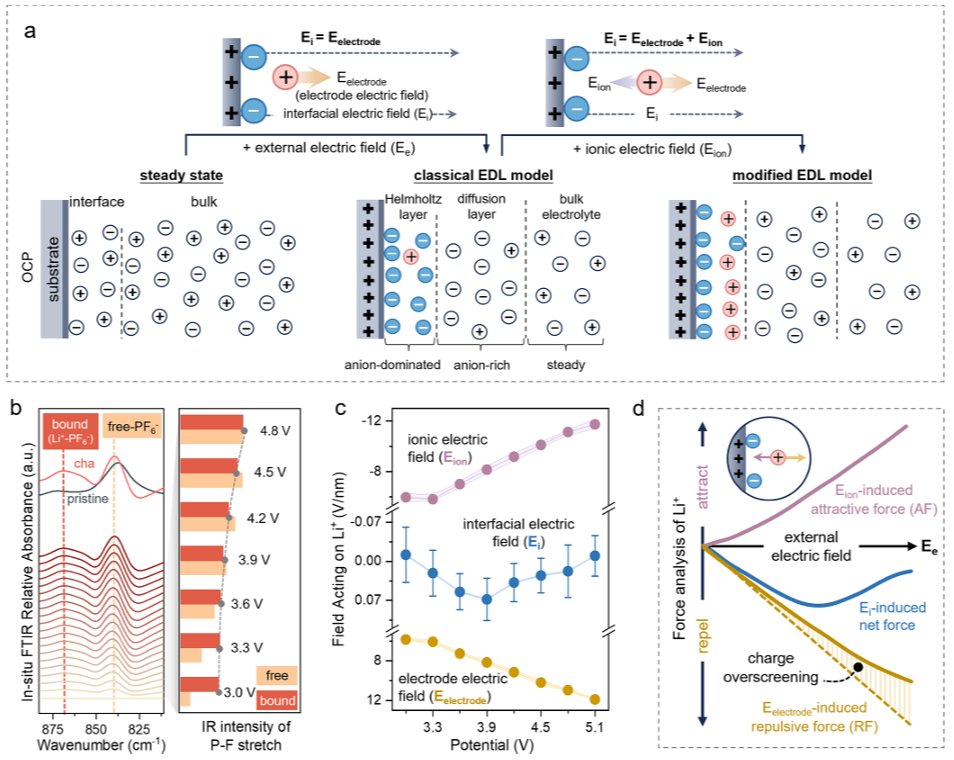
图1.界面电场和作用于界面Li+的净力的动态演变。(a)二维平面界面上的EDL模型图。左图展示了OCP下的EDL,代表了稳态电解质结构。中间和右图描绘了存在外部电场(Ee)时的EDL:(中间)经典模型;(右)引入离子电场(Eion)的改进模型。在经典模型中,界面电场(Ei)主要由电极(Eelectrode)产生的电场组成。引入Eion后,Ei为Eelectrode和Eion的矢量和。注意:金属电极与电解质之间的接触通常会导致OCP偏离零电荷点,从而导致界面电荷分布不均匀和界面电场(Ei)较弱。为简单起见,此处忽略OCP下的Ei,假设界面和本体电解质之间的物质分布相同。(b)线性扫描伏安法(LSV)期间P-F振动区域内Al表面的原位FTIR差异光谱(扣除OCP背景),表明随着施加电位的增加,界面处游离和配位PF6-的浓度均显著增加。深灰色线是原始电解质的红外吸光度。(c)模拟界面Li+所经受的电场强度与施加电位(3.05至5.1V)的关系。为了公平比较,统计区域限制在距电极表面0.8-1.8nm的范围内,因为在较高电位下,0-0.8nm区域内不存在Li+离子。(d)作用于界面Li+的力与电位关系的示意图。
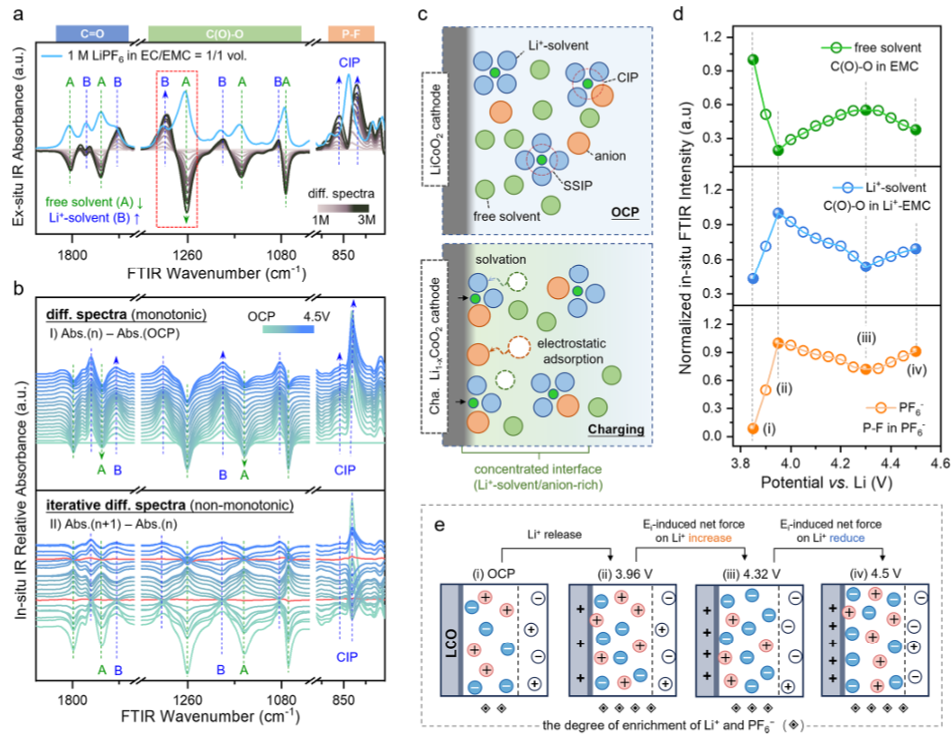
图2.LiCoO2正极表面界面电解质的多阶段演变。(a)不同浓度(1-3M)电解质的原位FTIR差异光谱,以1M光谱为背景。蓝线是1M电解质的FTIR吸收光谱。(b)(顶部)Li||LCO电池首次充电过程中LCO表面的原位FTIR差异光谱,其中OCP光谱作为背景减去。(底部)通过减去相邻光谱获得迭代差异光谱。(c)充电和OCP条件下界面的示意图比较。在充电过程中,Li+从正极释放并与吸附的溶剂分子配位,而阴离子静电吸附到LCO表面,从而形成浓缩界面。(d)Li||LCO电池中LCO表面电解质物质特征振动模式的电位相关红外吸收。(e)不同带电状态下界面离子分布示意图。白色菱形符号内含黑色小菱形,表示离子富集程度——符号越多,界面浓度越高。
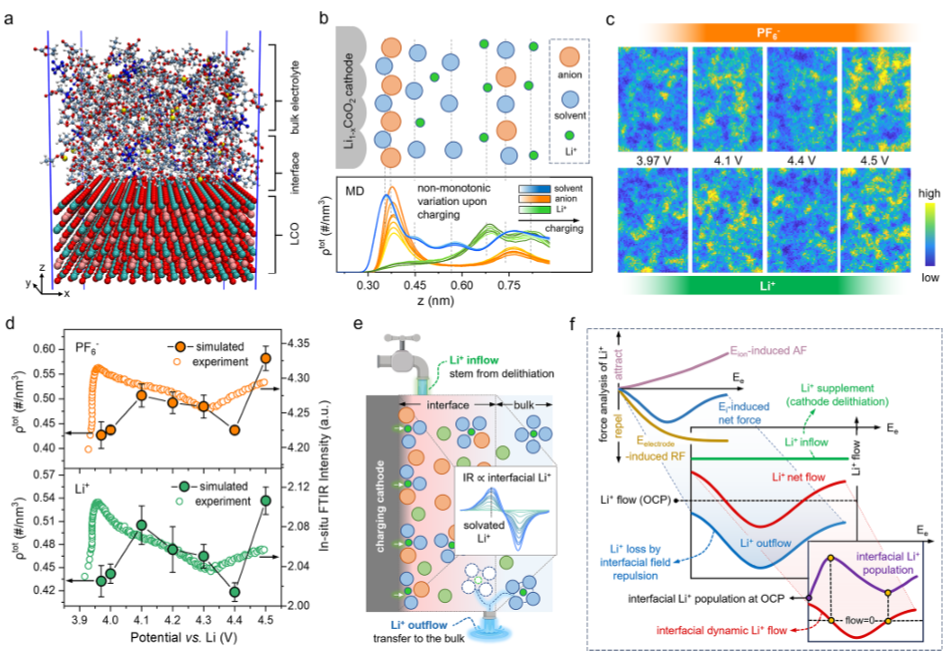
图3.充电正极动态界面演化模型的构建。(a)模拟系统中LCO-电解质界面的快照。(b)不同充电电位下界面电解质物质的数密度分布,以及相应的EDL示意图。虚线标记Li+(绿色)、PF6–(橙色)和EC/EMC(蓝色)密度分布中的峰值位置,颜色越深表示施加的电位越高。(c)模拟的不同电位下界面离子分布的示意图。(d)Li+和PF6–(实心圆)的电位相关累积数密度与原位FTIR吸收强度(空心圆)的关系。尽管转折点存在微小偏差(∼0.1V),但实验和模拟结果呈现出一致的宏观趋势:界面离子表现出多阶段的“富集-耗尽-再富集”过程。(e)界面Li+粒子数动力学的“储层模型”类比:正极脱锂(Li+流入)和电场驱动的Li+跨界面相迁移(Li+流出)共同决定了界面Li+粒子数。溶剂化Li+的原位FTIR吸收强度直接反映了界面Li+的这种变化。(f)决定界面Li+粒子数的界面“场-流”竞争模型示意图。界面电场通过静电排斥决定Li+的损失,而正极脱锂引起的Li+流动决定Li+的供应。这两个因素都通过控制界面处的动态Li+流动来决定实时Li+粒子数。
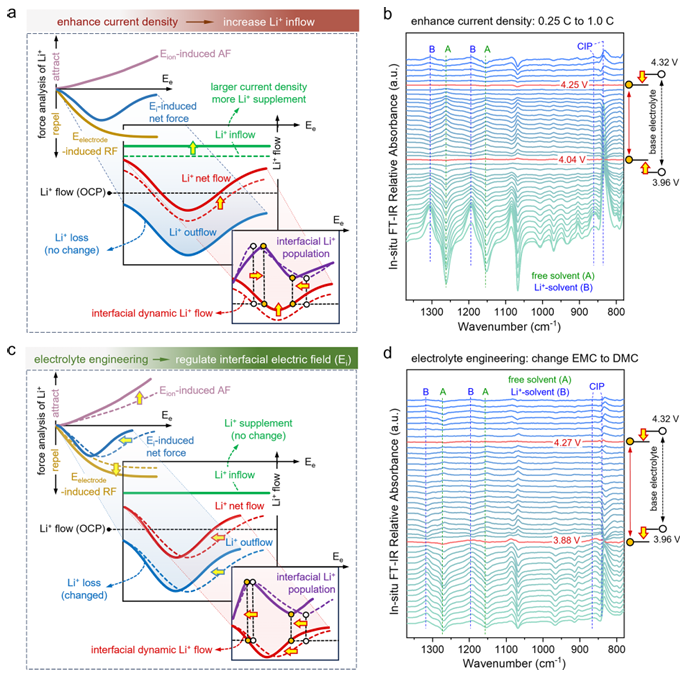
图4.电流密度和溶剂调节对界面Li+布居的影响。(a)示意模型分析和(b)原位FTIR展示了电流密度升高对界面Li+布居动力学的影响。较高的电流密度(1C)会增加Li+的流入并引起净流量向上移动。与基线(0.2C)相比,升高的电流会压缩拐点之间的电位窗口。(c)示意模型分析和(d)原位FTIR实验结果显示了不同介电常数溶剂的影响。用介电常数较低的溶剂DMC代替EMC,该溶剂为离子提供的静电屏蔽较弱,增强了阳离子和阴离子之间的相互作用。在低电位下,可解离的离子对较少,导致吸附的阴离子较少,对Li+的排斥力较强,导致第一个拐点电位提前移动。同时,在高电位下,离子间相互作用增强,增加了吸附阴离子对Li+的吸引力,降低了Li+所受的净力,导致第二个拐点电位提前发生偏移。
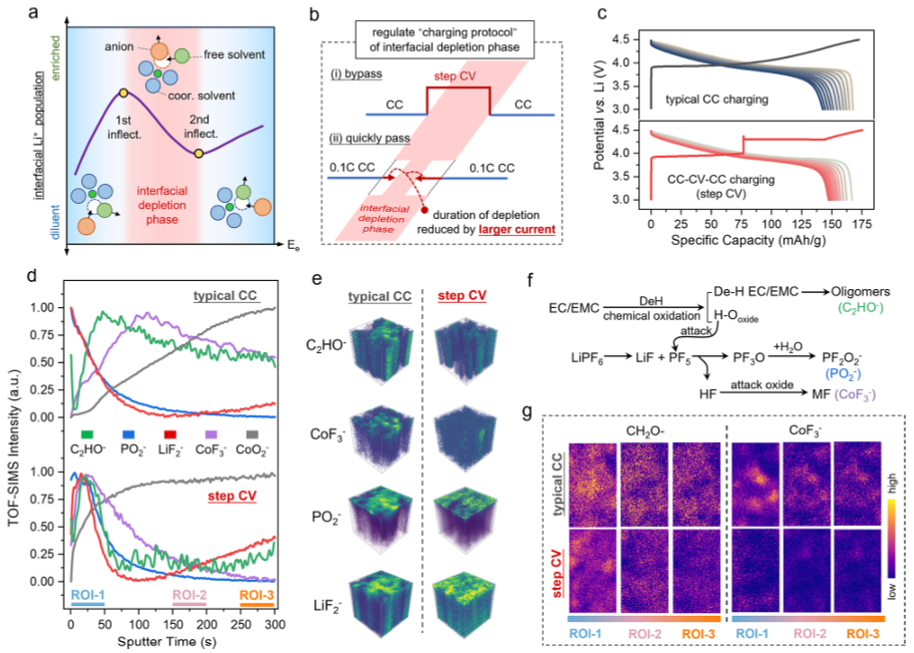
图5.充电方案调节调控CEI形成。(a)界面Li+布居三阶段演变示意图及相应的界面溶剂化构型。(b)基于降低界面耗尽区贡献的活化方案优化设计原理。(c)Li||LCO半电池在典型CC和阶梯CV活化后充电曲线及循环性能对比。分别对典型CC和阶梯CV活化3个循环后的CEI进行TOF-SIMS分析。(d)代表性无机和有机碎片的归一化深度剖面图;(e)不同活化方案后从正极提取的选定CEI碎片的相应3D渲染图。(f)LCO正极上电解液分解的氧化途径。(g)不同深度段中CH2O–和CoF3–碎片的TOF-SIMS化学映射图像。
研究结论
本研究结合原位光谱与分子动力学模拟,采用一种经典而强大的方法阐明了LiCoO2正极表面界面电解质结构的演变。我们揭示了充电过程中界面Li+粒子数的多阶段动态演变。随着施加电位的增加,这种演变遵循非单调的“富集-耗尽-再富集”趋势。为了使这些观察结果合理化,我们扩展了传统的GCS双电层模型,引入了离子电场和源自正极脱锂的动态Li+流。这使得我们能够系统地分析离子所受的力及其在电极-电解质界面处的分布。
基于通过原位红外光谱获得的界面电解质结构的动态信息,我们提取了界面Li+粒子数作为关键描述符,揭示了界面电场和离子流之间的动态竞争如何控制界面溶剂化的演变。具体而言:(1)界面电场(Ei)驱动的Li+损失主要受电极电场(Eelectrode)和离子产生电场(Eion)共同作用的影响,其中Eelectrode占主导地位。在充电过程中,界面电场强度随电位的升高先增大后减小。(2)脱锂产生的Li+流作为实时的Li+补充,该Li+流受充电电流控制,在恒流充电条件下保持不变。(3)在充电初期,Li+流主导界面Li+粒子的演变,导致界面阳离子和阴离子的富集,界面浓度增加。随着电位的升高,当Ei诱导的Li+迁移速率与Li+流补充速率达到动态平衡时,界面浓度的趋势发生逆转。随后,由电子对流 (Ei) 驱动的离子迁移占据主导地位,导致界面 Li+ 损失超过补充,并引发界面浓度下降。在充电后期,增强的阴离子吸附会削弱 Ei,降低排斥力,使 Li+ 在界面处重新富集,从而增加界面浓度。Li+ 损失(Ei 诱导的转移)与 Li+ 供应(受 Li+ 流入控制)之间的竞争最终决定了界面离子分布和电解质结构的动态演变。虽然该场流竞争模型有效地捕捉了界面 Li+ 动力学的关键静电方面,但界面处的化学因素(例如电解质分解和电极表面重构)仍可能影响电场和离子行为,这表明模型有进一步发展的潜力。
基于场流竞争模型,我们将电流密度、溶剂极性和施加电位对界面溶剂化动力学的调控作用分离。电流密度调节Li+的流动,而溶剂极性和施加电位则控制界面电场强度。在多阶段演化过程中,界面耗尽阶段Li+的粒子数减少,Li+–PF6–相互作用减弱,不利于形成富无机CEI。为了解决这个问题,我们设计了一种分阶段活化方案,该方案修改了耗尽阶段的充电方案,有效地减轻了其对CEI形成的不利影响。TOF-SIMS表征证实,该方法促进了富无机CEI的形成,抑制了电解质分解和过渡金属溶解,并显著提高了电池性能。除了优化活化方案外,场流竞争模型还启发了更广泛的界面设计策略,以缩短耗尽阶段。例如,可以采用具有优先吸附特性的电极表面涂层或添加剂来缩短Li+/PF6–耗尽界面状态的持续时间,从而为在高压工作条件下构建阴离子衍生CEI提供了一种通用方法。总体而言,本研究提出了调节界面溶剂化结构的新理论框架,同时为下一代电化学储能装置中的电解质工程和界面设计提供了有价值的指导。