
什么是d能带中心模型
在催化反应中,催化剂的催化活性同催化剂的电子构型和几何构型密切相关,由此发展出了许多表征催化剂活性与电子构型关联的理论和模型。目前主要使用的有前线分子轨道理论和d能带中心模型。在电催化反应中,包括了物种的吸附和电子的转移。其电子的转移难易可以通过催化剂的前线分子轨道来进行定性了解;而物种的吸附则可以通过d能带中心模型有效的模拟。
一.什么是d能带中心模型?
d能带模型是从前线轨道理论发展起来,由Nørskov和Hammer等提出,d能带模型主要用于异相催化和电化学中金属催化剂的模拟和分析。在异相催化和电化学中,分子和原子在金属催化剂或金属电极表面的吸附和成键非常重要,而d能带模型能够很好地描述由不同情况引起电子构型变化的金属催化剂与吸附物种吸附强度关系,进而根据d能带中心的变化来设计金属催化剂活性。
d能带模型之所以可以近似表征分子在金属表面的吸附能与离解的活化能,是因为对给定反应,吸附分子在不同金属表面的吸附和离解活化能近似地随着吸附物种能级和过渡金属d能带的耦合程度变化。
吸附物种-金属表面之间的成键主要由两部分组成:
ΔE=ΔE0+ΔEd
ΔE0是由吸附态价带与催化剂类似自由电子的s电子的耦合产生的键能,ΔEd是由吸附态价带与催化剂d电子的耦合产生的键能。其耦合过程如下图所示:
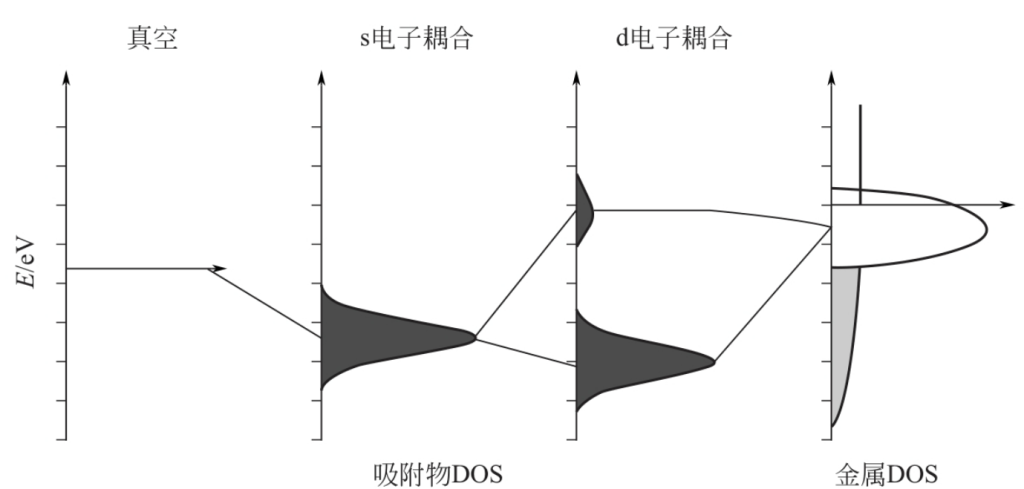
d带模型有两个基本假定:
一是ΔE0与金属种类无关。因为过渡金属的s带很宽,并且总是半满。尽管这并不是一个严格的近似,因为当金属颗粒足够小到s和p能级不能形成连续(在吸附物与金属耦合强度范围内)光谱时,该模型不成立。此外,当金属d能带对成键无贡献时,此模型也不成立
二是依照非自洽单电子能量的变化,预测d能带的贡献:
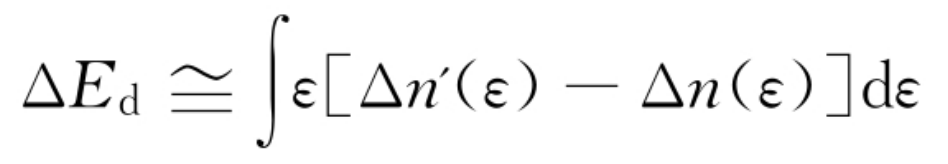
式中,Δn′(ε)和Δn(ε){是吸附质产生的能态密度,分别代表含有和不含与d带的耦合。
通常,吸附质能态与金属d能带的耦合依赖于以下参数:
a.吸附质能态的能量εa;
b.直接与吸附质接触的金属原子的d态密度nd(ε)
c.吸附质与表面态之间耦合矩阵单元。
对给定吸附分子的反应,吸附分子的εa和与金属表面态之间的矩阵单元是常数,只有nd(ε)变化。此时就可以只研究nd(ε)的第一力矩–d能带中心εd。d带中心εd是最简单的描述符,可以较好地描述吸附能的变化情况
必须注意的是,除了d能带中心外,相互作用能还与投影的d态密度nd(ε)的宽度和形状有关。然而,这些变量通常与d能带中心变化联系在一起,因此可以集中采用d能带中心描述。
为了说明此点,考虑到d带宽度[nd(ε)的二阶矩,W]会因为一些原因而减小。对固定的εd-εF,改变d能带宽度可能会使d电子数改变。但对给定种类的金属和体系,通过d能态能量的改变,使d电子数不变(如下图)。
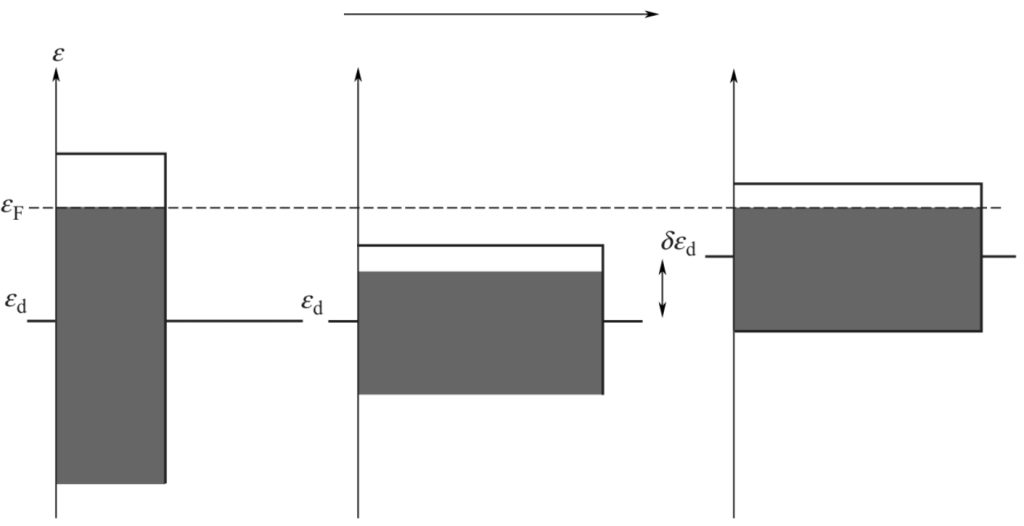
因此,可以通过d能带中心相对于Fermi位的变化εd-εF,来确定金属对分子物种的吸附能力。下式表示分子在金属表面的化学吸附同d能带中心的关系:

式中,f表示金属d轨道的填充情况;S是分子轨道同金属d轨道之间的重叠积分;V是分子轨道同金属d轨道之间的耦合矩阵。
此式表明化学吸附能,可以通过理论计算分子的HOMO和LUMO与金属的d能带中心的相互作用来预测。
当一个分子同金属表面相互作用时,其相互作用包括了电子从分子的最高已占分子轨道HOMO转移到金属表面(直接成键),以及金属表面电子迁移到分子最低未占分子轨道LUMO上(反馈成键)。对于过渡金属表面,电子的交换是通过金属d轨道的相互作用完成。在此模型中,d能带中心相对于Fermi能级的上移将导致大的化学吸附能,而下移则形成弱键。
二.d能带模型的应用
2.1金属表面构型的变化
特定的过渡金属的d带中心会因为金属配位数的变化,导致d能带中心的变化。例如:密堆积的(111)面Pt上原子配位数为9。在开放的(100)面,配位数为8,在阶梯或者(110)面上为7,在扭曲时配位数低至6。如下图所示,这些都导致d能带中心正移1eV。
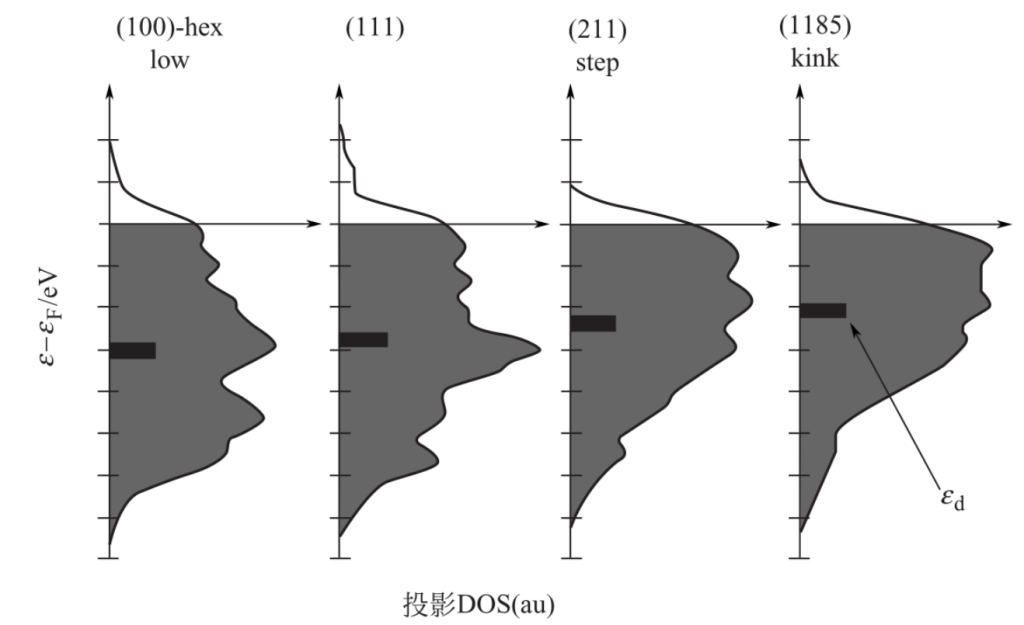
注意:d带中心与吸附物的种类,以及与金属表面相互作用的价带能级无关,由此说明了d带中心的普适性。
2.2合金的变化
合金效应同样可以通过d能带的变化来理解。如下图所示,图a为H2和O2吸附能vs.不同结构合金d带中心;图b为中间层金属对Pt/M/Pt合金d带中心的影响。
图b中以Pt(111)表面为例,该表面中有一系列的3d金属夹在第一层和第三层之间,该表面用来研究第二层金属对Pt(111)覆盖层反应性的影响。这种近表面合金,广泛地用于研究PEM燃料电池中的氧气还原催化剂。当所加入的第二层金属在周期表中越靠左(金属位于右面),表面Pt原子的d能态能量越低,随着d带中心离Fermi位越近,O和H物种与Pt的成键越强。
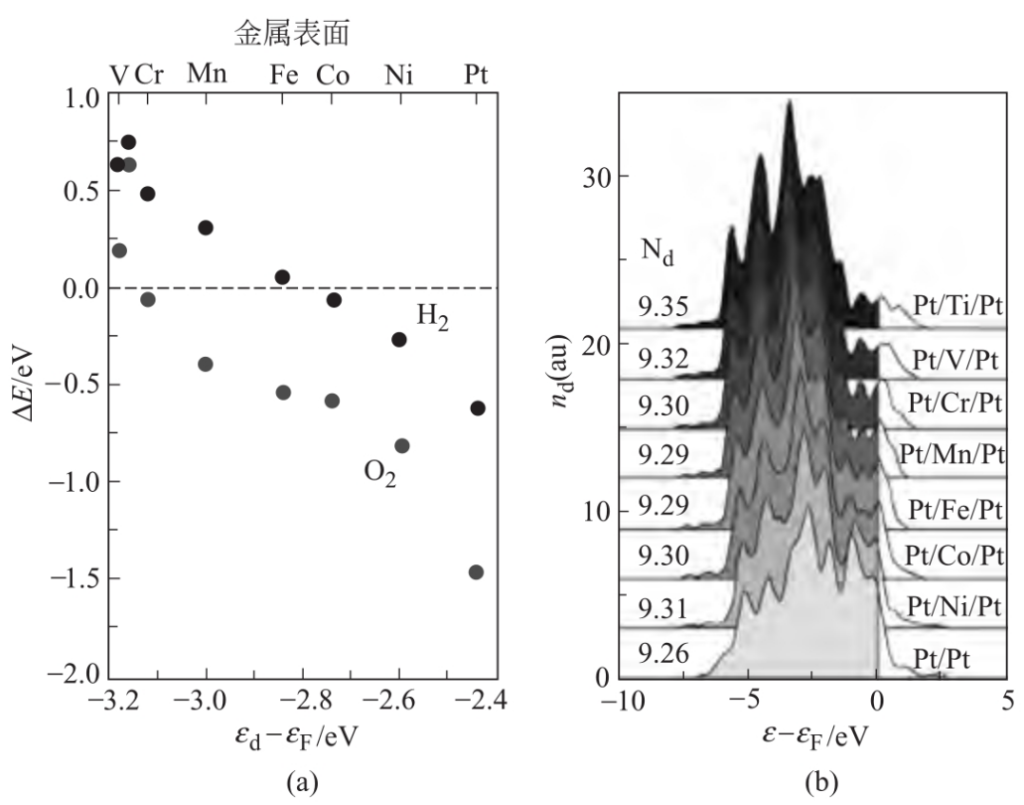
对近表面合金,表面Pt原子d轨道与第二层原子的杂化使d能带宽度发生变化。这种间接的相互作用同样可以解释为配位效应——表面原子的金属配体改变。
给定金属的反应活性可以通过沉积到另一种金属上进行改性,由此可以控制金属催化剂的反应活性。了解当金属沉积到其它金属上后其d带中心的变化,可根据需要选择有利的金属化合物。
d带中心理论:
d能带中心的能级高低决定了反键能带被电子填充的程度,从而决定了吸附成键的稳定性和强度。
从分子轨道理论出发,当吸附分子靠近金属表面的时候,吸附分子的轨道会和金属的s和d轨道发生作用导致能级分裂,形成的成建轨道会比原来的轨道能量更低更稳定,而同时形成的反键轨道会比原来能级能量还要高,不稳定。如果电子都填充到成键轨道之上,那么体系的整体能量是下降的。反之,反键轨道电子填充越多,结构越不稳定,中间体键合强度越弱。
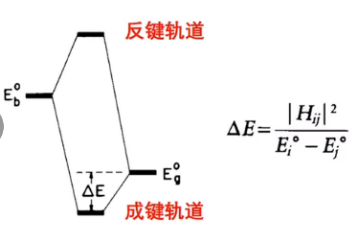
吸附作用之后,如果最终生成的反键态高于费米能级则有利于吸附,因为高能级的轨道上没有填充电子;如果最终生成的反键态低于费米能级则不利于吸附,因为高能级轨道上填充了电子。看金属对于分子的吸附是否稳定就看新生成的反键态的能级位置即可。
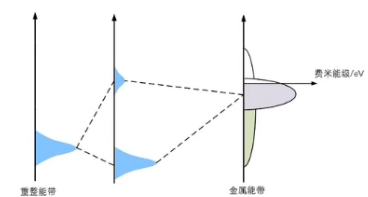
d-band中心越低,耦合产生的反键能带降低,更多部分的反键能带低于费米能级,从而被电子填充降低了键合的稳定性,吸附强度降低。